Research
Our research focuses on computer-aided approaches to identify functionally relevant genetic changes in disease and evolutionary adaptation as well as on the development of more sensitive methods in diagnostics (especially exome, genome and cell-free DNA sequencing). In general, our bioinformatics research spans the areas of sequence analysis, data mining, machine learning, and functional genomics.
Areas of Research
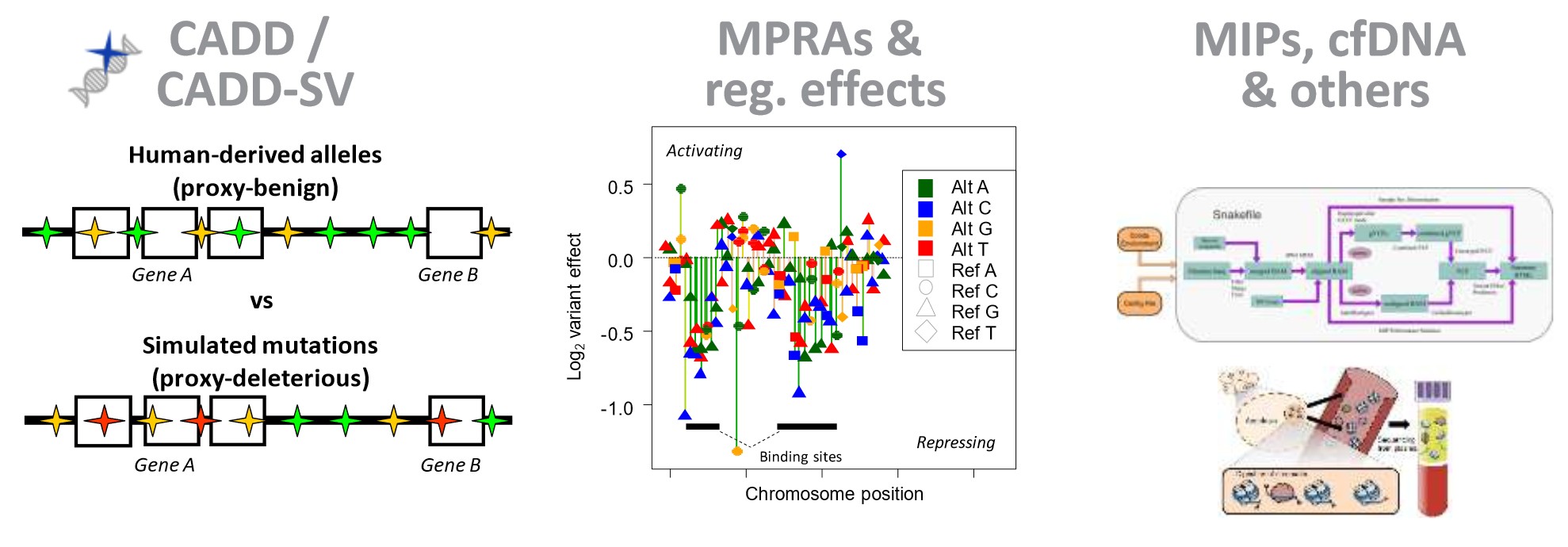
Genome-wide predictors of variant effects
We develop and maintain a widely used tool for assessing variant effects (Combined Annotation Dependent Depletion, CADD) that uses machine learning to integrate close to 100 different gene-based and genome-wide annotations. CADD was the first tool to predict variant deleteriousness for all possible single base pair changes in the human genome and also allows for the assessment of multibase and insertion/deletion changes. While there are many annotation and scoring tools for variants, most annotations tend to use a single type of information (e.g., sequence conservation) and/or are limited in scope (e.g., to missense changes). Therefore, a broadly applicable metric is needed that objectively weights and integrates diverse information. Combined Annotation Dependent Depletion (CADD) is a framework that integrates multiple annotations into one metric by comparing variants that survived natural selection to simulated mutations. CADD scores correlate with pathogenicity and allele frequency of both coding and non-coding variants.
In 2020, we extended the basic idea of CADD in a new project to also comprehensively assess structural variants (SVs) (CADD-SV). The interpretation of SVs is of great relevance due to recent technological advances in SV detection. CADD and CADD-SV can quantitatively prioritize disease causal and functional genetic variants across a wide range of functional categories, effect sizes, and genetic architectures, and can be used for variant prioritization in both research and clinical settings.
Functional genomic sequences and their organization
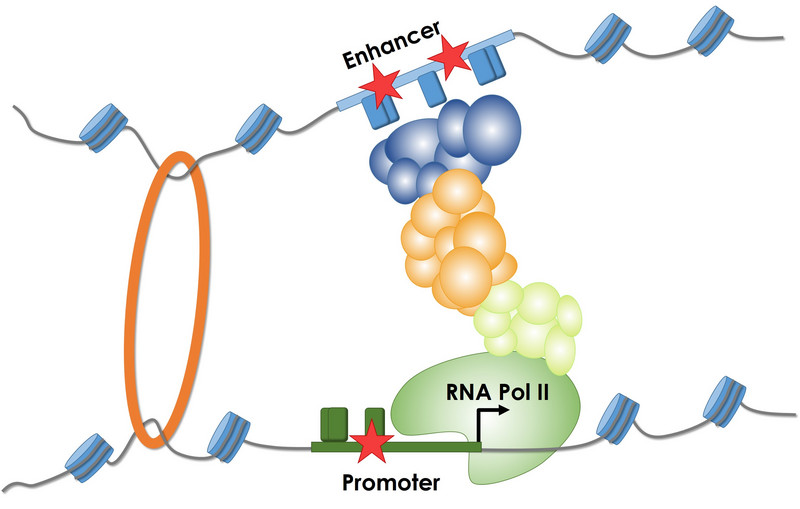
Regulatory sequences (promoters and enhancers) are involved in the control of gene expression. Mutations (stars) increase or decrease the binding of different proteins (transcription factors) to DNA sequence, thereby changing the amount of RNA which can impact certain phenotypes.
|
Regulatory sequences (e.g., promoters and enhancers) are involved in controlling gene expression. Sequence changes increase or decrease the binding of various proteins (e.g., transcription factors) to the DNA sequence, thereby altering the amount of RNA that is being synthesized, which can affect certain phenotypes. We are collaborating with the labs of Nadav Ahituv and Jay Shendure to obtain and analyze experimental measurements of noncoding sequence activity, in particular from Massively Parallel Reporter Assays (MPRA) and CRISPR/Cas9 activation/inhibition assays (CRISPRi/a). Although the majority of all genetic alterations affect non-coding sequences and there is growing evidence of significant phenotypic implications and clinical relevance, alterations in these sequences remain less well understood than alterations in coding regions. In this area we are developing and supporting the Regulatory Mendelian Mutation (ReMM) score. Furthermore, we use experimental data to derive computational models (e.g., gapped-kmers, CNNs and deep learning) of regulatory sequence effects with the aim of contributing to a better understanding of regulatory sequence function and to integrate them into genome-wide variant assessment. Here you can have a look at the effects of variants in 20 regulatory elements published in our saturation mutagenesis manuscript. |
Supporting the development of molecular assays and diagnostics
In addition, we develop pipelines to support analysis, data interpretation and visualization of DNA sequencing applications. This includes developments in primary data processing (e.g. base calling, read merging/adapter trimming, quantification), but also support for the development of new experimental protocols. For example, we investigated sample cross-contamination in Illumina multiplex experiments and proposed a protocol that introduces indexes into both sequencing library adapters (now known as double or dual indexing). We also developed analyses (e.g. HemoMIPs) for the targeted investigation of certain DNA regions (sequence enrichment/targeted sequencing). In other studies, we look at cell-free DNA (cfDNA) and show that nucleosome and transcription factor binding affect DNA fragmentation, thereby providing insights into cell type composition and pathological conditions.